








Servicios Personalizados
Articulo
 pdf en Inglés
pdf en Inglés Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Permalink
PermalinkLatin American applied research
versión impresa ISSN 0327-0793
Lat. Am. appl. res. v.32 n.2 Bahía Blanca abr./jun. 2002
Platinum monolithic catalyst for SO2 abatement in dust-free flue-gas from combustion units
E. Alvarez, J. Blanco*, J. Otero De Becerra1, J. Olivares Del Valle2 and L. Salvador2
* Instituto de Catálisis y Petroleoquímica, CSIC., Cantoblanco, 28049 Madrid, SPAIN.
1 Departamento de Combustión y Gasificación, CIEMAT, 28040, Madrid, SPAIN.
2 Departamento de Ingeniería Química y Ambiental, ESII, 41092 Sevilla, SPAIN.
Abstract — A catalytic oxidation process of traces of SO2 to SO3 for the treatment of dust-free flue-gas from power stations and some special waste incinerators has been developed. The catalyst was prepared by impregnation of platinum precursors on alumina/silicates-based material. The behavior of this industrial sized honeycombtype catalyst was studied at lab and pilotplant scale. The evolution of SO2 conversion and some physico-chemical properties of the catalyst with time in operation were examined. The 7-8 wt.% of sulfate deposition on the fresh catalyst had no influence on the catalytic activity at steady state conditions. The catalyst showed SO2 to SO3 conversion value of 80-90 vol.% operating at a temperature of 450ºC and space velocities of 3000-6150 h-1 (NTP).
Keywords — SO2 oxidation, industrial size, honeycomb-type, dust-free gas
I. INTRODUCTION
Nowadays, one of the tasks in environmental protection is directed towards the development of technologies for flue-gas SOx removal, where further reduction in its emission levels will be required in accordance with the future application of more stringent regulations.
Emission control techniques for the combustion of coal and other fossil fuels have been developed through improvements in pre, post and in-combustion treatments. In postcombustion methods, the most widespread procedure uses wet lime/limestone scrubbers and spray dryers for flue-gas desulfuration; latterly, the catalytic oxidation of sulfur dioxide to sulfur trioxide and further sulfuric acid production has emerged as an alternative, which could avoid the usual problematic disposal of the calcium sulfates produced from the conventional methods. In these catalytic systems, a useful SO3 byproduct is obtained from which various commercial products may be marketed, such as sulfuric acid and ammonium sulfate (Borio and Kingston., (1993); Blumrich and Engler, (1993)). Obviously, catalysts produced for commercial application should require dust-free flue-gas conditions.
The process might also be suitable for SO2 removal from flue-gas produced by combinedcombustion of coal and waste, such as tanned leather residue incineration. In Spain, tanned leather residues have been estimated as 104 T per year produced from shoe and leather accessories factories. The average content of chromium is about 4 wt.% as Cr2O3 while the sulfur content is around 2 wt.%, although this value can widely vary depending on the tanned leather. The chromium oxidation state in leather is Cr (III) with very low mobility in the environment and low toxicity for living organisms. Nevertheless, the large volume of the leather residues is giving rise to several problems as the storage capacities of the deposits are reached and the weathered chromium salts restrict dumping alternatives. The action of water and air produces the slow conversion of Cr (III) to Cr (VI) which could be lixiviated and transported away from the stored residues. The hexavalent chromium is a common toxic pollutant in soils and waters. Its action on human beings covers allergic contact dermatitis and cancers. Taking into account these considerations, incineration could be the best solution to get rid of this waste material. The combined-combustion of leather residues and coal is also an interesting alternative.
In the incineration of leather residues, the generated ashes, enriched in chromium (the content varies between 30-60 wt.%, depending on raw material and incineration conditions), can be used in ceramics, including decorative ones, to manufacture pavements and bricks, etc. During the incineration process, additional drawbacks arise as the leather residues contain significant sulfur content. The addition of limestone to retain the SO2 emissions produces the chromium (VI) release. The total oxidation of Cr (III) to Cr (VI) takes place if limestone is fed into the reactor, in the range of temperature between 450-600°C. In this case, the catalytic oxidation of SO2 containing flue-gas and subsequently sulfuric acid recovery might be a suitable solution.
In dust-free flue-gas treatment, the abrasion effect is minimized and noble-metal impregnated catalyst is currently used in the abatement systems (Truex et al., 1992). In the SO2 reduction field, some reports described noble-metal catalysts for SO2 oxidation, where the SO3 obtained is further converted to sulfuric acid. As an example in the DeSONOx process (Blumrich and Engel (1993); Ohlms, 1993), the previously dust cleaned flue-gas, was passed through a precious metal monolithic catalyst; afterwards, the SO3-rich flue-gas was cooled and sulfuric acid (70 wt.%) condensed. In this work, short pieces of 15 cm length were manufactured and plugging problems between layers by dust arose.
In the development of Pt-impregnated honeycomb catalyst for SO2 oxidation, the objective of seeking a sulfate-proof support has given rise to a certain number of publications and patents. For alumina, Ziolek et al.(1996) reported that SO2 alone is chemisorbed at 350°C. Moreover, the SO2 adsorption on alumina mainly takes place via the hydroxyl groups, giving rise to hydrogen sulfite species (Mohammed-Saad et al., 1995). When O2 is present, significant quantities of SO2 are oxidized to SO3 and subsequently stored on the alumina. When Pt is added, the SO2 oxidation process is catalyzed by the noble metal and SO2/SO3 are adsorbed on different alumina sites (Summers, 1979). Li- Dun et al., (1991) pointed out two types of sulfate species on the alumina having different environments: free sulfates and sulfates associated with the carrier.
No information about SO2/SO3 adsorption on sepiolite or aluminium silicates behavior has been reported.
The role of the platinum loading and dispersion on the carrier for the SO2 oxidation (Xue et al., 1996) has also been pointed out. When a Pt/alumina catalyst is used, the catalytic activity is not strongly affected by the Pt particle size and, there is a maximum loading percentage at which the excess of noble metal does not enhance the performance of the catalyst (Matsuda et al., 1982).
In this study, a Pt-impregnated on alumina/silicates-based support catalyst has been developed and tested both at laboratory and pilot-plant scale. The pilot plant has been designed to test the industrial sized catalyst at real operational conditions in order to evaluate the performance of the material. The evolution of SO2 conversion and some physico-chemical properties of both fresh and used catalyst were examined.
II. EXPERIMENTAL SECTION
A. Catalyst Preparation
The preparation method followed four steps: the carrier conformation, drying and calcination, Ptspecies impregnation and subsequent reducing.
The material of the support was prepared by mixing the corresponding quantities of three different components: a) a boehmite with an average particle size of 5 µm, b) a natural α- sepiolite containing ca. 36% of impurities, mainly other clays and calcium carbonate with an average aggregate-size of about 11 µm, and c) a natural high-purity aluminum silicate with an average particle size of 9 µm. The support composition (by weight) was 2/2/1 for alumina, α-sepiolite and aluminum silicate, respectively.
The three components of the support were thoroughly dry mixed before adding water. Kneading of this slurry was performed until homogeneous dough was obtained. The material was conformed using an industrial-scale extruder. Parallelepiped monoliths of 10x10x100 cm external dimensions were obtained with square cells of 4x4 mm open-cell and 1 mm wall thickness. In the case of operation in low-dust conditions as a result of a malfunction of the dust collector, the 1 m length would avoid the interlayer plugging problems, which arise when shorter monoliths are used.
The conformed structures were dried at 110°C and calcined at 500°C for 4 hours causing 12% shrinkage. The bulk density of the prepared carrier was 1.3 g cm-3 and the mechanical strength was 119 kg cm-2.
The impregnation of this support was carried out using a 600 ppm aqueous H2Cl6Pt solution. The impregnation process was performed immersing one monolith each time. A movable cage holding the material and allowing a constant vertical motion was built. This way, the Pt concentration inside the monolith channels was kept constant. The average time for each impregnation was 15 minutes. After each impregnation process, the released liquid volume was replaced trying to maintain the initial Pt concentration of 600 ppm. The evolution of Pt concentration in the solutions was monitored by Atomic Absorption technique. After impregnation, the monoliths were treated in ammonia flow at 400°C for 4 hours to produce the total Pt species reduction. XPS analysis of the prepared catalyst gave an average percentage of 0.12 wt.% of metallic platinum.
B. Characterization Techniques
N2 adsorption/desorption isotherms at 77 K were determined using a Micromeritics ASAP 2300-10D. Samples of the catalyst were outgassed overnight at 140°C to a vacuum of < 10-4 torr to ensure a dry clean surface. Surface area determinations were made by application of the BET equation. Total pore volume was determined using a CE Instrument Pascal 140/240 porosimeter. Samples were also dried at 140°C overnight. The mechanical strength of the monolithic catalyst was determined using a Chatillon LTCM Universal Tensile Compression and Spring Tester with a cylinder test head of 1 mm diameter. The test head was positioned over a channel wall and the pressure slowly increased until rupture of the wall was caused; the average of ten consecutive measurements was taken to ensure the accuracy of the result.
Thermal Gravimetric Analysis (TGA) determination was made using a thermobalance Perkin Elmer TGA 7 connected to a Fisons MD 800 mass spectrometer using a heating ramp of 10 °C min-1 within the temperature range of 30-950°C in 20 cm3 min-1 helium flow rate.
Platinum concentration analysis was carried using a Perkin Elmer 3030 Atomic Absorption (AA) Spectrophotometer. Standard AA conditions for Pt were 265.9 nm wavelength, 0.7 nm slit and air/acetylene flame.
X-Ray photoelectron spectroscopy (XPS) analysis of Pt was carried out using a Perkin Elmer spectrometer model Phi 5400, with a monochromatic Mg source (1254 eV), operating at 12 kV and 20 mA. The samples were outgassed under a vacuum of 10-8 bar before placing in the analysis chamber.
A Varian 3400 model Gas Chromatograph coupled to a flame photometric detector was used to monitor the inlet/outlet SO2 concentration in the lab-scale experiment.
The inlet/outlet SO2 and O2 measures in the pilot plant were performed using an ESSA ML9850 model Ultraviolet Fluorescence analyzer.
The EPA Method (Method 8, 1990) was selected for SO3 sampling procedure; the collected sulfuric acid mist was measured by the barium-thorine titration method.
III. RESULTS AND DISCUSSION
A. Lab-Scale Catalytic Activity Test
A sample from the industrial sized monolith was selected to test the catalytic performance at lab-scale conditions. The geometric dimension of the monolith piece corresponded to a 6-open-channel structure of 25 cm length. The catalytic activity measurement was carried out in a reactor working close to the isothermal axial profile, where carborundum was used to fix and isolate the catalyst. In order to avoid the entrance of SO3 to the GC, a 98 wt.% sulfuric acid trap was placed at the reactor outlet. The operation conditions are summarized in Table 1.
| Table 1. Operation conditions (NTP) for the lab-scale test. |
The SO2 conversion to SO3 increased with time in operation and reached a constant value of 90 vol.% after the first 6 hours. No deactivation of the catalyst was observed during the following 25 hours of the test, and the conversion was kept stable at the indicated value.
B. Pilot plant catalytic activity test
A scheme of the pilot plant installation is shown in Fig. 1. The main body is a catalytic reactor containing a basket with the package of honeycomb-type monoliths arranged vertically. The prismatic basket was built to provide two separate layers of 5x4 monoliths of 9.5x9.5x100 cm with a maximum catalyst load of ca. 330 liters. The dimensions of the reactor and coupling accessories to the main gas duct assured an isokinetic gas flow at the entrance of the catalytic bed. The head unit was a gas generator constituted by a gas-oil combustion chamber with a capacity of 200-500 STD m3 h-1 of simulated flue-gas in the range of 350-500ºC.
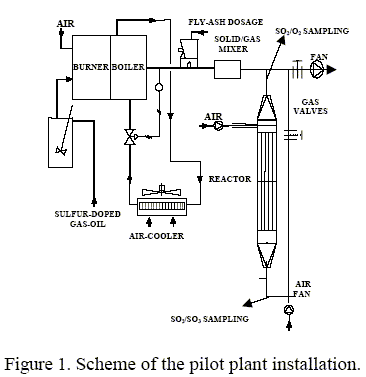
Varying the amount of carbon sulfide added to the gas-oil as the doping agent, the desired SO2 concentration in the flue gas was obtained. The air supplier valve placed in the burner controlled the variation of O2 concentration.
The catalyst placed in the prismatic holder occupied 151 liters total volume with a crosssection of 0.17 m2. Gas generation was about 430-470 Nm3 h-1 in order to achieve a GHSV of around 3000 h-1. The pressure drop through the catalytic bed was in the range of 21-24 mm of H2O m-1. The main operational parameters are summarized in Table 2.

In addition to the above-indicated compounds, the flue gas contained CO2, CO, NOx (ca. 300 ppm) and unburned hydrocarbons (ca. 600 ppm).
Two glass heated lined probes were connected upstream and downstream of the reactor to withdraw an aliquot for the SOx analysis. The temperature was recorded at the top and bottom of the reactor. A further thermocouple was located at 15 cm depth along a selected central monolith channel from the top of the reactor.
In Fig. 2, the temperature of the thermocouple inside the monolithic channel, the total flow and the SO2 inlet and SO2/SO3 outlet are plotted against the time in operation. Only data from the first 20 hours were plotted. The reaction system reached the stationary state after 13 hours, with SO2 to SO3 conversion values of about 80% until the run stopped after 40 hours in operation. Stable flow, temperature and O2 concentration values were observed over the test.
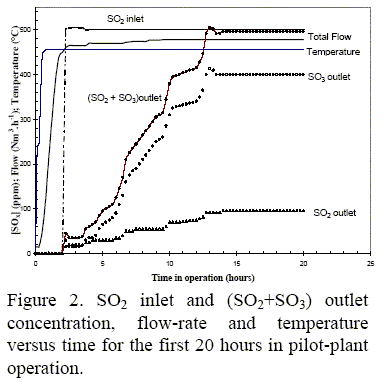
From Fig. 2 it can be clearly seen that the catalyst underwent a sulfating process during the first 13 hours in operation as also observed in the lab-scale test. After this step, the catalyst presented stable behavior with an 80 vol.% SO2 to SO3 conversion, lower than that observed in the lab-scale experiment.
Textural properties comparison between the fresh and used catalysts has been performed in order to evaluate the effect of the SOx uptake during the pilot plant run. The fresh catalyst presented a BET surface area (SBET) of 159 m2 g-1 and a total pore volume (Vp) of 0.53 cm3 g- 1. After reaction, the SBET and Vp decreased to 123 m2 g-1 and 0.48 cm3 g-1, respectively. Pore size distributions of both fresh and used catalysts are plotted in Fig. 3.
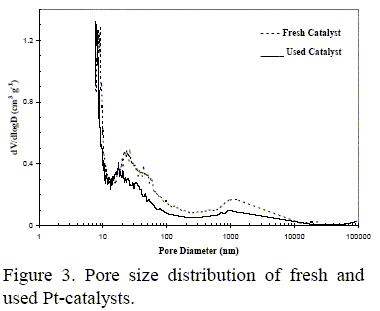
The fresh catalyst presented a mesopores with an average pore diameter centered at ca. 10 nm and a broad range between 15-100 nm in the mesoporosity zone; in the macropore range a broad peak within 0.6 and 10 µm pore diameter would correspond to interparticulateThe fresh catalyst presented a mesopores with an average pore diameter centered at ca. 10 nm and a broad range between 15-100 nm in the mesoporosity zone; in the macropore range a broad peak within 0.6 and 10 µm pore diameter would correspond to interparticulate space. For the used catalyst, it can be seen that pores with a diameter greater than 30 nm were affected by the sulfate deposition. Otherwise, sulfating process did not markedly alter the mesopores with diameter below 30 nm existing in the fresh catalyst. Moreover, the slight decrease of the BET surface area values indicated that the sulfatization was mainly taking place in the range of meso and macropores.
Thermal gravimetric (TG) and mass spectrometry (MS) combined techniques were used to monitor weight loss and gas released with temperature. The results corresponding to the fresh and used catalysts are shown in Fig. 4.
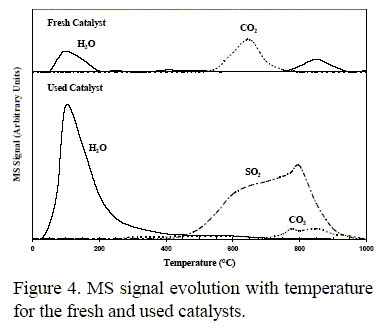
For the fresh catalyst the 6.3% total weight loss between 20 and 950°C mainly corresponded to H2O and CO2. In the used sample, TG analysis showed between 20 and 400°C an 8% weight loss due to H2O release. Between 500-950°C second range presented a 7.5% weight loss, which was mainly related to the sulfite/sulfate decomposition.
With respect to the water signal, in the fresh catalyst, different H2O or hydroxyl groups were removed as the temperature was increased. Two types of water molecules can be distinguished. The absorbed water leaving the catalyst between 80-200°C and hydroxyl groups probably coming from the alumina at 800°C.
In the used catalyst, the MS results showed the water signal between 20-400°C. The used catalyst became more hygroscopic as a consequence of the sulfite/sulfate groups deposition. No water MS signal at about 800°C was recorded, therefore, the hydroxyl groups surface were also altered.
With respect to CO2, in the fresh catalyst, the carbonates from sepiolite were removed between 500 and 750°C. No CO2 in the used sample in the range of 500-700°C was detected and the carbonate decomposition took place at higher temperature (700-900°C) than that in the fresh sample. The CO2 peak in the used catalyst started to show up when the SO2 peak reached its maximum value. This CO2 might come from the unreacted carbonate, which would have been covered by the sulfate species.
With respect to the sulfate deposition, at least two types of sulfates were removed from the used catalyst. The SO2 starting to be released at ca. 500°C might correspond to the sulfates produced from the interaction between silicates and SO3. A further SO2 peak showing up when the temperature reached ca. 700°C might be assigned to the aluminum sulfate previously formed.
As the TG analysis indicated, the total estimated amount of sulfite/sulfate uptake by the catalyst was 7.5%. This value may be compared with that obtained from the pilot plant results. A simple calculation from the SO2 inlet, SO2+SO3 outlet concentrations and total flow data found that 6% (as SO3) would be the amount of deposited on the catalysts.
The mechanical strength of the monolithic catalyst was not altered by the 6-7 wt.% of deposited sulfate, maintaining the fresh sample value of 119 kg cm-2.
IV. CONCLUSIONS
The fresh catalyst adsorbed up to 7-8% (as SO3) of SOx. Afterwards, a stable 90-80 vol.% SO2 to SO3 conversion was achieved at laboratory and pilot-plant scales respectively.
The textural characteristics were not strongly affected by the sulfate deposition. BET surface area and total pore volume were not dramatically altered. Mesopores of less than 10 nm were unchanged, but the decrease of wider mesopores and macropores was the more significant effect.
The difference of 10 points in the conversion value between the laboratory and pilot-plant runs might be mainly assigned to the presence of water vapor and unburned hydrocarbons presenting the pilot-plant gas stream.
The catalyst presents an acceptable catalytic activity with no observed poisoning action by SOx along with adequate mechanical properties. Therefore, this feasible Ptimpregnated monolithic catalyst based on silicates/alumina might be applied in the SO2 emission reduction from dust-free flue-gas.
Acknowledgments
The authors are thankful to CICYT, ECSC and CSE-ENDESA-OCICARBON for their financial support. The authors gratefully acknowledge the support from the ECSC project (Nº 7220-ED/080).
We are also grateful to Dr. Yates for his help in obtaining and discussing the textural data and to Dr J. Pérez-Pariente and Dr.E. Sastre for the TPD-MS analysis. Also, we wish to thank Dr. C. Knapp for his valuable scientific support.
REFERENCES
1. Blumrich, S. and B. Engler, "The DESONOX/ REDOX-process for flue gas cleaning", Catalysis Today 17, 301-309 (1993).
2. Borio, D. C. and W.H. Kingston, "Efficient and cost effective NOx and SO2 removal from flue gas with the SNOXTM process", Processing and Utilization of High-Sulfur Coals V, 451-465 (1993).
3. Li-Dun, A., D. You-Quan, X. Tian-Cun, X. Hong-Bing and L. Sheng-Li, "The mechanism of sulfur-poisoning of Pd(Pt)/Al2O3 catalysts and their regeneration", Bartholomew C.H. & Butt J. B. (eds). Catalyst Deactivation, 557-580 (1991).
4. Matsuda, S., A. Kato, T. Mori, T. Kumagai, Y. Hishinuma, H. Akimoto and F. Nakajima, "Process for treating flue-gas", U.S. Patent 4,350,670, (1982).
5. Method 8- "Determination of sulfuric acid mist and sulfur dioxide emissions from stationary sources", Environmental Protection Agency. Pt. 60, App. A, Method. 8. 40 CFR Ch. I (7-1-90 Edition)(1990).
6. Mohammed Saad, A.B., O. Saur, Y. Wang, C.P. Tripp, B.A. Morrow and J.C. Lavalley, "Effect of sodium on the adsorption of SO2 on Al2O3 and on its reaction with H2S", J. Phys. Chem. 99, 4620-4625 (1995).
7. Ohlms N., "The DESONOX process for flue gas cleaning", Catalysis Today 16, 247-261 (1993).
8. Summers J.C., "Reaction of sulfur oxides with alumina and platinum/alumina", Environmental Science & Technology 13(3), 321-325 (1979).
9. Truex, T.J., R.A. Searles, D.C. Sun, "The opportunity for new technology to complement platinum group metal autocatalysts", Platinum Metals Review 36(1), 2-11 (1992).
10. Xue, E., K. Seshean and J.R.H. Ross, "Catalytic control of diesel engine particulate emission: studies on model reactions over a EuroPt-1 (Pt/SiO2) catalyst", Applied Catalysis B: Environmental 11, 65-79 (1996).
11. Ziolek, M., J. Kujawa, O. Saur, A. Aboulayt and J.C. Lavalley, "Influence of sulfur dioxide adsorption on the surface properties of metal oxides", Journal of Molecular Catalysis A: Chemical 112, 125-132 (1996).
Received: February 24, 2000.
Accepted for publication: April 26, 2000.
Recommended by Subject Editor G. Meira.
